Technology innovation for crop improvement
KeyGene is the go-to research company for the development and application of breakthrough technology innovation. We do this for and with our shareholders and other partners, for crop improvement in for instance vegetables, fruits, ornamentals, and industrial field crops.
Technology innovation
We develop and apply breakthrough technology innovation for crop improvement, for and with partners around the globe.
Crop improvement
We use proprietary technology innovation to support partners around the globe to boost their crop improvement.

9 April 2024
Advancing Smart Breeding for Resilient Agriculture: A Conversation with CropXR Managing Director Hedwich Teunissen
Hedwich Teunissen, Managing Director of CropXR, shares insights into the institute's misison, science and collaborative approach.

6 March 2024
Recent evolution of Fusarium TR4 strain has increased diversity in ‘virulence’-DNA of the fungus
Paper: Evolution of Fusarium TR4 strain has increased diversity in ‘virulence’-DNA of the fungus
22 February 2024
Studying nuclei of individual pollen grains is feasible now
A collaboration between Takara Bio and KeyGene has made it possible to carry out single-nucleus RNA sequencing (snRNA-seq) successfully.
Take a look at our work

Accelerated breeding of impatiens resistant to downy mildew
KeyGene supported Ball Horticultural in the USA to reveal, understand and utilize the genetic variation at hand within their germplasm, for accelerated breeding of impatiens varieties resistant to downy mildew

Hybrid rice for farmers in India
Experts from KeyGene and Bioseed, part of DCM Shriram in India, together developed rice lines that can be used by Biodseed to breed hybrid rice varieties that meet the demands…
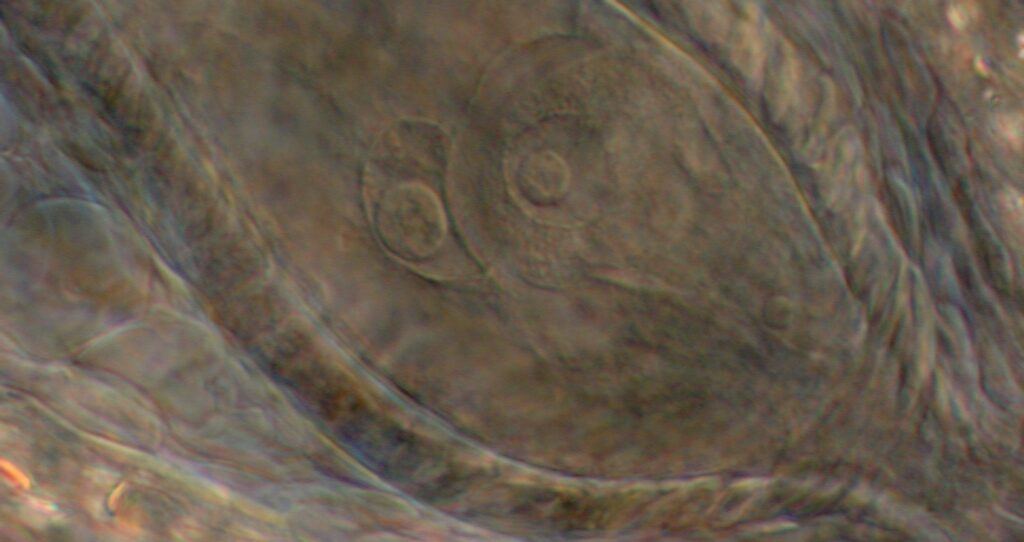
Genetics unraveled crucial to making crop plants produce clonal seeds through apomixis
Thanks to over 15 years of fundamental research at KeyGene, breakthroughs were achieved in understanding and using genes crucial to develop crop plants that produce 'clonal' seeds through apomixis